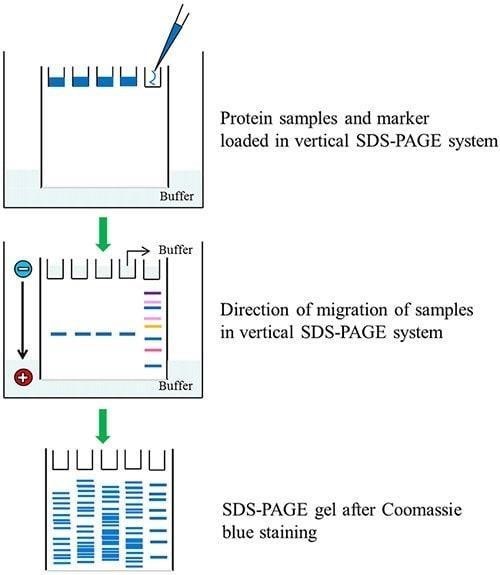
Introduction
Electrophoresis is the process of migration of charged molecules in response to an electric field. The rate of migration depends on the net charge, size and shape of the molecule, the voltage gradient of the electric field E, and the frictional resistance of the supporting medium f, which impedes their movement. Proteins have a net charge at any pH other than their isoelectric point (pI), thus when placed in an electric field, proteins will migrate towards the electrode of the opposite charge.
This principle is used to separate molecules of differing charges. Electrophoresis in acrylamide gels is referred to as Polyacrylamide gel electrophoresis (PAGE). Polyacrylamide gels which were first used for electrophoresis by Raymond &Weintraub (1959) are chemically inert and particularly stable. By chemical copolymerization of acrylamide monomers with cross linking reagent N-N’-methylene bisacrylamide a clear transparent gel which exhibits little endosmosis is obtained.
The polymerization of acrylamide is an example of free
radical catalysis and is initiated by the addition of Ammonium per sulfate and
a catalyst N,N,N’N’- Tetramethylenediamine(TEMED).TEMED catalyses the
decomposition of the persulfate ion to give a free radical.
S2O8 2- +e- →SO42- +SO4 –
If this free radical is represented as R![]() (where the dot represents an
unpaired electron) and M as an acrylamide monomer molecule than the
polymerization can be represented as follows.
(where the dot represents an
unpaired electron) and M as an acrylamide monomer molecule than the
polymerization can be represented as follows.
R![]() +M→ RM
+M→ RM ![]()
RM![]() +M→RMM
+M→RMM![]()
RMM![]() +M→RMMM
+M→RMMM![]()
In this way long chains of acrylamide are built up being
crosslinked by introduction of bisacrylamide forming a mesh like structure in
which the holes of the mesh represent the pores. Overall protein mobility
through polyacrylamide gel is proportional to the pore size which is a function
of both the acrylamide concentration (%T) and that of bisacrylamide crosslinker
(%C.).In general the pore size is inversely proportional to %T.
The proteins may be run in denaturating conditions in presence
of SDS or in native condition devoid of denaturants called as native- PAGE of
proteins. In native or non-denaturing gel electrophoresis SDS is not used and
the proteins retain their native structure and enzymatic activity. Although the
resolution is not as high as that of SDS-PAGE but the technique is useful when
the enzymatic activity of a protein need to be assayed following
electrophoresis. The migration of proteins in non-denaturating gel is due to
both the net charge and the size of the protein.
SDS-PAGE is the most commonly used gel electrophoretic system
for analyzing proteins. This method is based on the separation of proteins
according to size and can also be used to determine the relative molecular mass
of proteins. SDS is an anionic detergent which binds strongly to and denatures
proteins to produce linear polypeptide chains. On average one SDS molecule will
be present for every two aminoacids. The presence of β-mercaptoethanol assists
in protein denaturation by reducing all disulfide bonds. The detergent binds to
the hydrophobic region of the denatured protein in a constant ratio of about
1.4g of SDS/gm of protein. The protein- SDS complex carries net negative
charges, hence move towards the anode and the separation is based on the size
of the proteins. Most SDS-PAGE gels are cast with a molar ratio of
Bisacrylamide:Acrylamide of 1:29 which has been shown empirically to be capable
of resolving polypeptides that differ in size by little as 3%.
The Polyacrylamide gel is cast as a separating gel topped by a
stacking gel. The stacking gel has properties that cause the proteins in the
sample to be concentrated into a narrow band at the top of the separating gel.
This is achieved by utilizing differences in ionic strength and pH between the
resolving buffer and the stacking gel and involves a phenomenon known as
isotachophoresis. The stacking gel is of high porosity and buffered with
Tris-cl buffer at pH 6.8, whereas separating gel contains high percentage of
acrylamide and is cast in Tris-cl buffer at pH 8.8. The upper (and lower)
electrophoresis buffers contain Tris at pH 8.3 with glycine as counter ion.
Stacking principle: Glycine at pH 6.8 of the stacking gel
remain in neutral zwitterionic form with only a fraction 1% in the negative
glycinate form. This prevents glycine to be an effective carrier of current.
The Cl- ions remain effective current carriers at pH 6.8 and migrate rapidly
towards the anode. The SDS-coated protein molecules and dye which have charge
to mass ratio >glycine but less than that of Clmust now migrate to carry the
electrophoresis current behind the Cl- and ahead of the glycine. There is only
a small quantity of protein-SDS complexes so they concentrate in a thin band
sandwiched between the cl- ions and the glycine molecules at the interface
between stacking and separating gels.
The higher pH of the separating gel favours ionization of
glycine, carrying a higher charge to mass ratio than that of the proteins. Now
the newly formed glycinate ions move faster than the proteins with mobility
approaching that of the cl- ions. The negatively charged protein-SDS move
according to their relative mobilities and are separated by the sieving effect
of the separating gel according to size. The high mobility of the tracking dye
assures that it will migrate faster than the proteins.
Protein resolved in the gel can be stained with either Coomaassie brilliant blue or with silver stain. Silver staining is the most widely used high sensitivity staining method which is reported to be 100 times more sensitve than Coomassie blue with a detection limit about 0.1-1ng of protein. Coomassie blue are electrostatically attracted to charged groups on the protein, forming strong dye:vprotein complexes that are further augmented by vanderwaals forces, hydrogen bonding and hydrophobic bonding. On the other hand selective reduction of silver ions to metallic silver at gel sites occupied by proteins is the principle of silver staining. It depends on the differences in the oxidation-reduction potentials in the sites occupied by the proteins in comparison with adjacent sites in the gel that do not contain proteins.
Requirements
30% acrylamide: weigh 29g acrylamide, 1g N, N –
methylene bis-acrylamide. Add 60 ml warmed deionized water and heat to 37 ℃.
Add deionized water to make a final volume of 100ml; filter; Then we have 30%
(w / v) acrylamide stock solution; Acrylamide and bis-acrylamide were
transformed slowly into acrylic acid and double acrylic acid during storage, so
the pH of the solution should be no more than 7.0 and it should be placed in a
brow bottle at 4 ℃.
10% sodium dodecyl sulfate (SDS): weigh 10g SDS and
90ml deionized water; heat to 68 ℃ and add a few drops of
concentrated hydrochloric acid until the pH becomes 7.2; then water to 100ml;
after the whole processes, we have 10% (w/v) SDS.
Stacking gel buffer (1mol / L Tris-HCl pH 6.8):
dissolve 12.12g Tris in 80ml deionized water. Adjust the pH to 6.8 with
concentrated hydrochloric acid; add deionized water to 100ml and store at 4℃.
Resolving gel buffer (1.5mol / L Tris-HCl pH 8.8):
dissolve 18.16g Tris in 80ml deionized water; adjust the pH to 8.8 with
concentrated hydrochloric acid; add deionized water to 100ml; store at 4 ℃.
10% ammonium persulfate (AP): ammonium persulfate
provides the free radical necessary for the catalysis of the Polymerization of
Acrylamide and Bis-acrylamide; Use deionized water to prepare a small amount of
10%
(w/v) solution and store at 4 ℃. Since ammonium persulfate will
decompose slowly, it should be freshly prepared every other week.
TEMED (N, N, N, N – tetramethylethylenediamine): by
catalyzing ammounium persulfate to form free radicals, TEMED accelerated the
polymerization of acrylamide and bis-acrylamide. Since TEMED only functions in
a free base form, the polymerization reaction would be inhibited when the pH is
low.
Tris- glycine electrophoresis buffer: weigh 15.1g
Tris and 94g glycine; Dissolve in 900ml deionized water; then add 50ml 10%
(w/v) SDS and deionized water to 1000ml. Dilute 5-fold when using. The final
concentration would be: Tris, 25mmol/L; glycine, 250mmol/L; SDS, 0.1% and the
pH of the buffer is 8.3.
Protein Stain: 0.1% Coomassie brilliant blue R250 in
50% methanol, 10%glacial acetic acid. Dissolve the dye in the methanol and
water component first, and then add the acetic acid. Filter the solution
through whatmann filter paper. (Note:Coomassie brilliant blue is harmful by
inhalation or ingestion. Wear appropriate gloves & safety glasses while
handling)
Destaining solution: 10% methanol, 7% glacial acetic
acid 10. Protein sample 11. Standard Protein molecular weight markers.
Polyacrylamide gel electrophoresis tank and electrophoresis
power supply.
Transfer pipette and tip, etc.
Procedure
- Clean the internal surfaces of the gel plates with methylated
spirits, dry, and then join the gel plates together to form the cassette, clamp
it in a vertical position.
- In an Erlenmeyer flask or disposable plastic tube, prepare the separating gel by mixing the following: (NOTE1)

- Degas this solution under vacuum for about 30sec. (NOTE2)
- Add 14µl of TEMED and gently swirl the flask to ensure even
mixing. Using a Pasteur pipette transfer this separating gel mixture to the gel
cassette carefully down one edge. Continue adding this solution until it
reaches a position 1cm from the bottom of the comb that will form the loading
wells.
- To ensure that the gel sets with a smooth surface very
carefully run distilled water down one edge into the cassette using a Pasteur
pipette.
- While the separating gel is setting prepare the 4% stacking
gel solution. Mix the following in a 100ml Erlenmeyer flask or disposable
plastic tube. 0.6M Tris-Hcl, pH6.8 1.0ml Stock acrylamide 1.35ml Water 7.5ml
10%SDS 0.1ml Ammonium persulfate (10%) 0.05ml Degas this solution under vacuum
for about 30 sec
- When the separating gel has set, pour off the overlaying
water. Add 14 µl of TEMED to the stacking gel. Pour the stacking gel solution
directly onto the surface of the polymerized resolving gel. Immediately insert
a clean Teflon comb into the stacking gel solution, being careful to avoid
trapping of air bubbles. Place the gel in a vertical position at room
temperature and allow to set for 20min. Preparation of samples and running the
gel:
- About 10µl of protein sample and 5µl of sample buffer are
mixed by vortexing. The sample is than heated for 5min at 95-100°C to denature
the proteins. The sample is than kept in ice (Note3)
- After polymerization is complete, remove the Teflon comb. Rinse out any unpolymerised acrylamide solution from the wells using electrophoresis buffer and assemble the cassette in the electrophoresis tank. Add Tris-glycine electrophoresis buffer to the top and bottom reservoirs. (Note: Donot prerun the gel before loading the samples, since this procedure will destroy the discontinuity of buffer system.)
- Load up to 5-10µl of each of the samples (unknown and
standard) in a predetermined order into the wells.
- Connect the electrophoresis apparatus to the power pack (the
positive electrode should be connected to the bottom buffer reservoir), and
pass a current of 30mA through the gel (constant current) for large format
gels, or 200V (constant voltage) for minigels (Biorad). The gel is run until
the bromophenol blue reaches the bottom of the resolving gel. This will take
2.5-3.0h for large format gels (16µm x 16µm) and about 40min for minigels (10µm
x7µm)
- (Safety care: Always turnoff & disconnect the power supply
before removing the lid)
- Dismantle the gel apparatus, pry open the gel plates; remove
the gel, discard the stacking gel, and place the separating gel in stain
solution.
- Staining should be carried out with shaking, for a minimum of
2h at room temperature. Destain the gel by soaking it in the methanol:acetic
acid solution on a slowly rocking platform for 4-8 hrs.
- After destaining, store the gels in water containing 20% glycerol.
- The gel can now be used for immunoblotting to determine the
protein sample.
NOTE
- Typically15% polyacrylamide gels are used for separating
proteins of molecular mass in the range of 100,000-10,000 kd.However,a protein
of 150,000for example would be unable to enter a 15% gel.In this case, a large
pored gel(eg a 10% or 7.5% gel) would be used.
- Degassing helps prevent oxygen in the solution “mopping up”
free radicals and inhibiting polymerization.
- β-mercaptoethanol is essential for disrupting disulphide
bridges in proteins. However exposure to air decreases the reducing power of
β-mercaptoethanol. Thus it should be prepared fresh.
- Destain solution needs to be replaced at regular intervals
since a simple equilibrium is quickly set up between the concentration of stain
in the gel and destain solution after which no further destaining takes place.
- It is generally accepted that a very faint protein band
detected by Coomassie brilliant blue, is equivalent to about 0.1µg(100ng) of
protein
- Data Analysis Label each lane on the photograph of your gel:
The molecular weight of the unknown protein can be determined by running
calibration proteins of known molecular weight on the same gel run as the
unknown protein.
- A standard curve is constructed that plots relative mobility (Rf) versus Log mol weight Rf =(distance migrated by protein/distance migrated by tracking dye)
- The Rf of the standard protein is calculated and plotted on the graph. The Rf value of the unknown protein is calculated by measuring the distance each protein band migrated (Measure from the bottom of the well to the middle of each band) and the distance the tracking dye migrated in each lane. Using the standard curve, the molecular weight of the unknown protein is determined.
Result

Reference
- Russell, D. W., & Sambrook, J. (2001). Molecular cloning: a laboratory manual (Vol. 1). Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Sambrook, J., & Russell, D. W. (2006). SDS-polyacrylamide gel electrophoresis of proteins. CSH Protoc, 1(4).